Terapie genowe zarejestrowane w Europie oraz Stanach Zjednoczonych
Terapie genowe zarejestrowane w Europie oraz Stanach Zjednoczonych
Terapia genowa to metoda leczenia chorób genetycznych polegająca na wprowadzeniu do komórek prawidłowej kopii genu, którego defekt jest przyczyną choroby. Celem terapii genowej może być również włączenie lub wyłączenie funkcji danego genu czy też wprowadzenie do organizmu dodatkowego genu o działaniu terapeutycznym. Terapie genowe dają nadzieję na lepsze i dłuższe życie pacjentom chorującym na choroby rzadkie lub trudne w leczeniu. Artykuł przedstawia zestawienie zatwierdzonych terapii genowych w Europie i Stanach Zjednoczonych.
Dynamiczny rozwój biologii molekularnej przyczynia się do lepszego poznawania mechanizmów leżących u podstawy wielu chorób o podłożu genetycznym. Ponadto znaczny postęp w technologii transferu genów zaowocował rozwojem terapii genowej. Terapie genowe są obecnie wykorzystywane do leczenia często złożonych i trudnych do wyleczenia jednostek chorobowych. Są one stosowane do regulacji, naprawy, zastępowania, dodawania lub usuwania sekwencji kwasu nukleinowego, a sam efekt terapeutyczny, zapobiegawczy i diagnostyczny jest w bezpośrednim związku z sekwencją rekombinowanego kwasu nukleinowego, którą zawiera produkt lub w bezpośrednim związku z produktem, który powstaje na podstawie tej informacji genetycznej. Do grudnia 2023 r. zatwierdzono łącznie aż 19 terapii genowych w Europie oraz Stanach Zjednoczonych. Na szczególną uwagę zasługuje miniony rok 2023, gdyż był on przełomowy w dziedzinie terapii genowych ze względu na zatwierdzenie pierwszej terapii genowej, która jest oparta na przełomowej technologii CRISPR/Cas9.
WPROWADZENIE
W najbliższej przyszłości terapia genowa, jako nowatorskie badanie medycyny molekularnej, może mieć znaczący wpływ na zdrowie człowieka. Niegdyś pojęcie „terapia genowa” było utożsamiane z wprowadzaniem prawidłowej kopii genów bezpośrednio do komórki z nieaktywnym lub też uszkodzonym genem. W wyniku przeprowadzenia powyższej modyfikacji możliwe stało się przywrócenie syntezy określonego białka, dzięki czemu terapia genowa mogła być wykorzystywana w leczeniu chorób uwarunkowanych monogenetycznie, a zatem jednostek chorobowych, w których mutacja dotyczy wyłącznie pojedynczego genu, jak np. mukowiscydoza [1]. W ostatnich latach zakres terapii genowej uległ rozszerzeniu i obecnie zaczyna znacznie rewolucjonizować podejścia terapeutyczne. Warto także wspomnieć, iż technologia rekombinacji DNA zapewnia możliwość przeprowadzania badań genetycznych płodu i poradnictwa genetycznego. Dzięki terapii genowej udowodniono również słuszność koncepcji wielu chorób, w tym chorób dziedzicznych, takich jak dziedziczne niedoboru odporności, lizosomalne choroby spichrzeniowe, chorób o podłożu neurologicznym, chorób hematologicznych oraz chorób nowotworowych. Ponadto postępy w terapii genowej są obecnie wykorzystane w celu korygowania dziedzicznych zaburzeń genetycznych, takich jak hemofilia, mukowiscydoza i hipercholesterolemia rodzinna, a także innych chorób, takich jak nowotwór, choroba Alzheimera oraz choroba Parkinsona czy chorób zakaźnych, takich jak choroba wywołana przez ludzki wirus upośledzenia odporności (ang. human immunodeficiency virus; HIV) [2].
HISTORIA TERAPII GENOWEJ
Terapia genowa jest dziedziną nauki polegającą na dokonywaniu określonych zmian w genomie człowieka. Zmiany te obejmują zarówno wymianę, jak i edycję zmutowanych genów, a także wprowadzenie kopii genów do komórek w celu przywrócenia normalnej funkcji białek. Terapia genowa jest stosowana w chorobach uwarunkowanych genowo w celu uzyskania efektów terapeutycznych, wyleczenia lub poprawy stanu pacjenta. Zanim przedmiotowa terapia zaistniała przeprowadzono liczne eksperymenty oraz włożono wiele wysiłku, aby ugruntować i udoskonalić tę dziedzinę nauki do takiego poziomu, jaki osiągnęła ona obecnie. Pod koniec lat 70. i na początku lat 80. XX wieku słusznie przewidywano, iż techniki subklonowania genów ssaków do plazmidów prokariotycznych i bakteriofagów będą prekursorem technik terapii genowej u ludzi. Równolegle prowadzone badania biologii onko-retrowirusów ptasich i mysich doprowadziły do opracowania wektorów retrowirusowych, które od połowy lat 80. XX wieku były powszechnie stosowane w laboratoriach jako niezwykle cenny sposób przenoszenia genów do komórek ssaków. W związku z powyższym pod koniec lat 80. XX wieku przygotowano grunt pod pierwsze próby transferu genów z udziałem ludzi, które obejmowały badanie znakowania genowego limfocytów naciekających guz (ang. tumor-infiltrating lymphocytes; TIL) przeprowadzone przez grupę dr Stevena Rosenberga w 1989 r. [3-5]. Natomiast grupa badawcza dr Ro Michaela Blaese’a w 1990 r. przeprowadziła pierwszą udaną terapię genową u pacjenta z ciężkim złożonym niedoborem odporności (ang. severe combined immunodeficieny; SCID) spowodowanym brakiem deaminazy adenozyny (ang. denosine deaminase; ADA) [6]. Jest to kluczowy enzym dla prawidłowego funkcjonowania limfocytów T krwi obwodowej, odpowiedzialnych za odpowiedź immunologiczną. Niedobór przedmiotowego enzymu ADA przyczynia się do rozwoju częstych zakażeń o ciężkim przebiegu, co w następstwie zazwyczaj prowadzi do śmierci pacjenta jeszcze przed ukończeniem 2 roku życia. Przebieg przedmiotowej terapii genowej obejmował wyizolowanie limfocytów T, a następnie wprowadzenie do nich prawidłowej kopii uszkodzonego genu. Tak zmodyfikowane limfocyty zostały ponownie wprowadzone do ludzkiego organizmu. Dzięki zastosowaniu wspomnianej terapii genowej w ciągu kilku lat zaobserwowano wzrost stężenia ADA u pacjenta. Ponadto jego stan zdrowia uległ znacznej poprawie [6, 7].
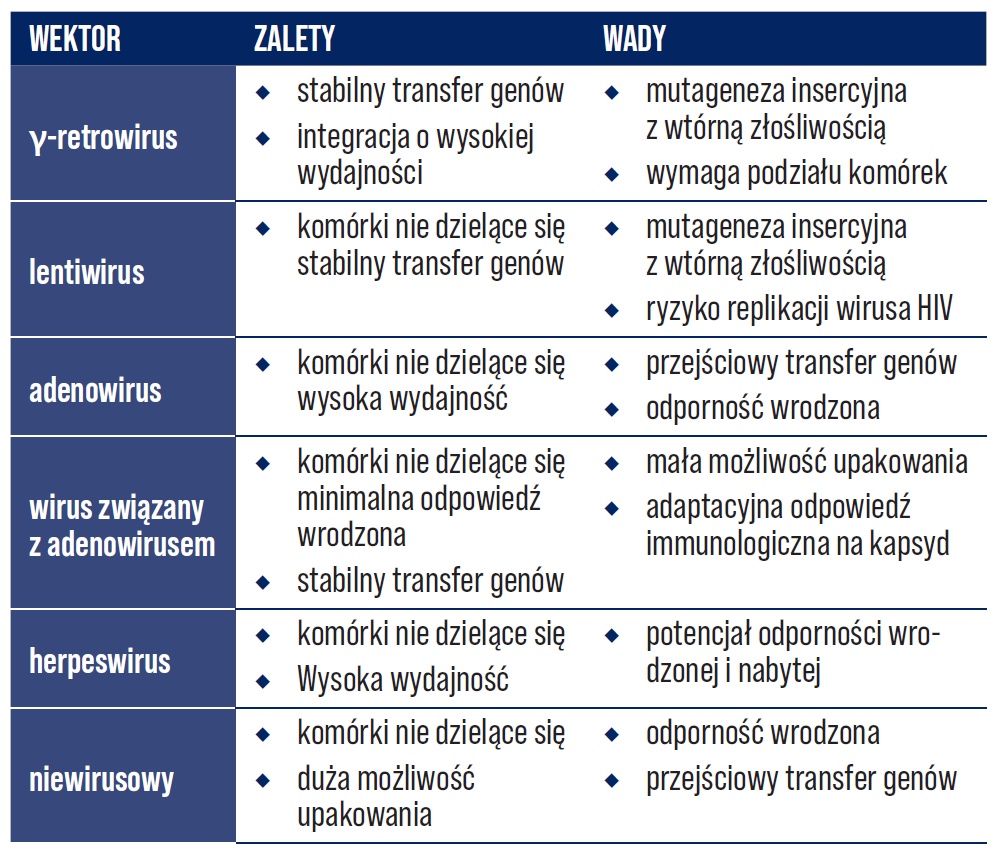
Od tamtej pory nastąpił znaczny wzrost liczby odkrytych genów ludzkich, które są bezpośrednio związane ze stanami chorobowymi, jak i niezwykły wzrost liczby dostępnych systemów wektorowych umożliwiających ekspresję genów w celach terapeutycznych. W kwietniu 1993 r. rozpoczęto prowadzenie terapii genowej wektorami rekombinowanego adenowirusa u pacjentów z mukowiscydozą [8], a 2 lata później rozpoczęto także pierwsze badanie z rekombinowanym wirusem związanym z adenowirusem (ang. recombinant adeno-associated virus; rAAV) także u pacjentów z mukowiscydozą [3, 9, 10]. Wkrótce potem rozpoczęły się badania na pacjentach z hemofilią B [3, 11]. Następnie na arenie klinicznej pojawiły się wektory lentiwirusowe oraz rekombinowane wirusy opryszczki [3, 12-16]. Każdy z czterech ostatnich wektorów jest zdolny do przenoszenia genów oraz ekspresji in vivo w niedzielących się komórkach. W latach 90. XX wieku w terapii genowej zaczęto również stosować różnorodne niewirusowe metody transferu genów, w tym liposomy kationowe, koniugaty DNA i wstrzykiwanie nagiego (ang. naked) plazmidowego DNA. Każdy z tych systemów wektorowych posiada zalety i wady, co zostało zobrazowane w Tabeli 1.
Tabela 1. Charakterystyka systemów wektorowych stosowanych obecnie w badaniach klinicznych [3].
RODZAJE TERAPII GENOWEJ
Terapię genową można podzielić na dwie grupy. Pierwsza z nich to terapia genową linii zarodkowej, tzw. terapia germinalna [2, 17]. W terapii genowej linii zarodkowej geny są wprowadzane bezpośrednio do komórek rozrodczych – plemników lub komórek jajowych, a także do komórek będących w początkowym stadium zarodkowym. Terapia ta nie jest obecnie powszechnie stosowana ze względów etycznych. Drugą grupę stanowi somatyczna terapia genowa, która obejmuje kompensacje skutków defektu genetycznego. Mechanizm ten polega na wprowadzeniu prawidłowej kopii genu objętego defektem, jak również na włączeniu bądź wyłączeniu funkcji danego genu. Ponadto możliwe jest również wprowadzenie dodatkowego genu, tzw. genu terapeutycznego. Natomiast w terapii genowej nowotworów wykorzystuje się geny, które niszczą komórki nowotworowe bądź prowadzą do zahamowania ich wzrostu [2, 7]. Somatyczną terapię genową można podzielić na dwie następujące kategorie: in vivo i ex vivo (Rycina 1) [2].
Rycina 1. Koncepcje terapii genowej in vivo i ex vivo Kaufmann K., Büning H., Galy A., Schambach A., Grez M. Gene therapy on the move. EMBO Mol Med. 2013, 5, 1644. https://www.ncbi.nlm.nih.gov/pmc/articles/PMC3840483/

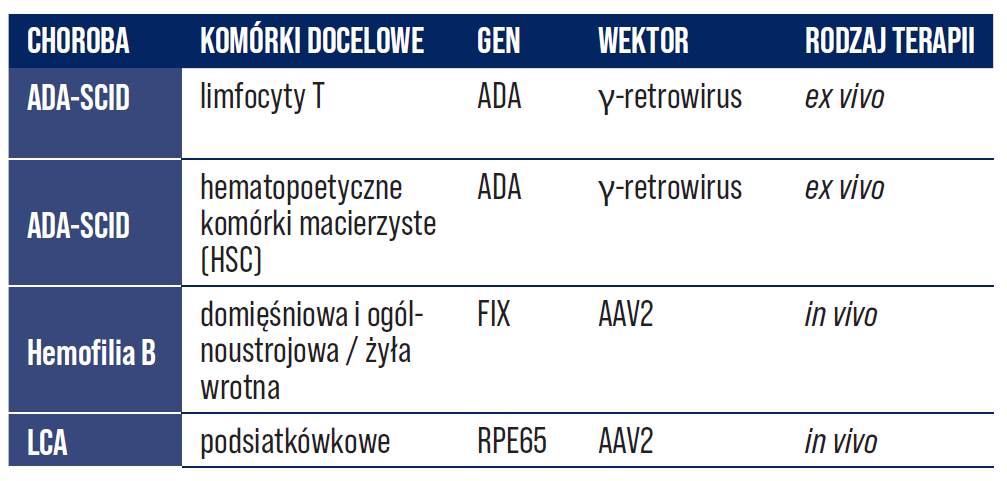
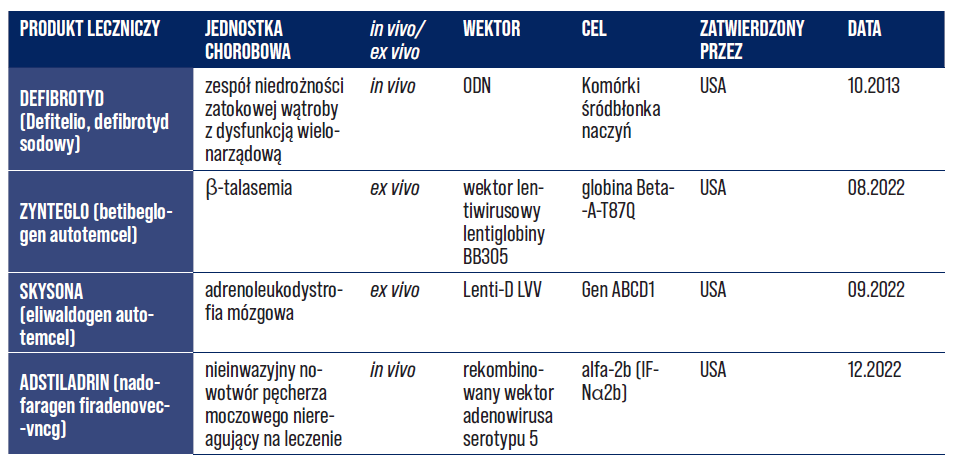
Terapia genowa ex vivo polega na pobraniu komórek od dawcy, transdukowaniu komórek docelowym genem i ostatecznie wprowadzeniu komórek do organizmu pacjenta (ryc. 1) [2, 18]. Terapia genowa przeprowadzana w warunkach in vivo sprowadza się do bezpośredniego wprowadzenia funkcjonalnych i prawidłowych genów do komórek pacjenta za pomocą różnych typów wektorów. W tabeli nr 2 zobrazowano niektóre z ważnych chorób leczonych z wykorzystaniem terapii genowej ex vivo i in vivo wraz z ich docelowymi komórkami, transgenami i wektorami [2, 18, 19].
Tabela 2. Potencjalne choroby wraz z uwzględnieniem komórek docelowych, transgenów i wektorów wykorzystywanych w terapii genowej in vivo lub ex vivo [2].
TERAPIE GENOWE – BIOLOGICZNE PRODUKTY LECZNICZE
W ostatniej dekadzie na rynek wprowadzono pierwsze zaawansowane terapie genowe. Terapie genowe to biologiczne produkty lecznicze. Ich składniki aktywne zawierają kwas nukleinowy (nośnik informacji genetycznej). Terapie genowe są wykorzystywane do regulacji, naprawy, zastępowania, dodawania lub usuwania sekwencji kwasu nukleinowego. Efekt zapobiegawczy, diagnostyczny i terapeutyczny wielu chorób, w tym m.in. zaburzeń genetycznych, nowotworów lub chorób przewlekłych, jest w bezpośrednim związku z sekwencją rekombinowanego kwasu nukleinowego, którą zawiera produkt lub w bezpośrednim związku z produktem, który powstaje na podstawie tej informacji genetycznej [20]. Do grudnia 2023 r. zatwierdzono łącznie aż 19 terapii genowych w Europie oraz Stanach Zjednoczonych (Tabela 3, 4, 5).
Tabela 3. Zestawienie produktów leczniczych terapii genowej zatwierdzonych w UE i USA [33].
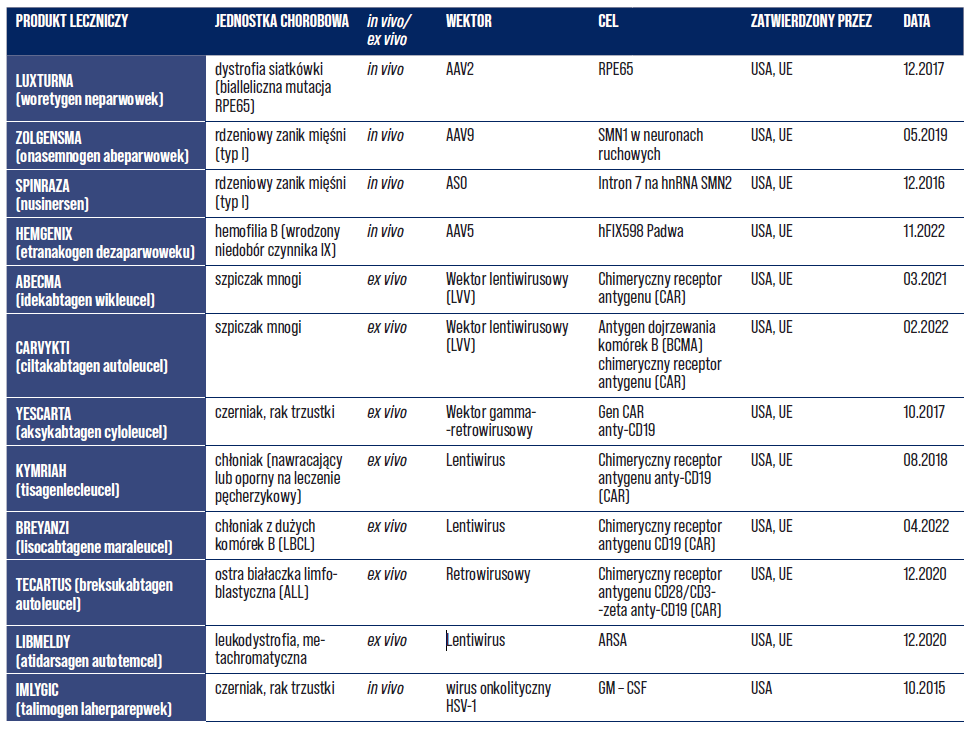
Tabela 4. Zestawienie produktów leczniczych terapii genowej zatwierdzonych wyłącznie w UE [33].
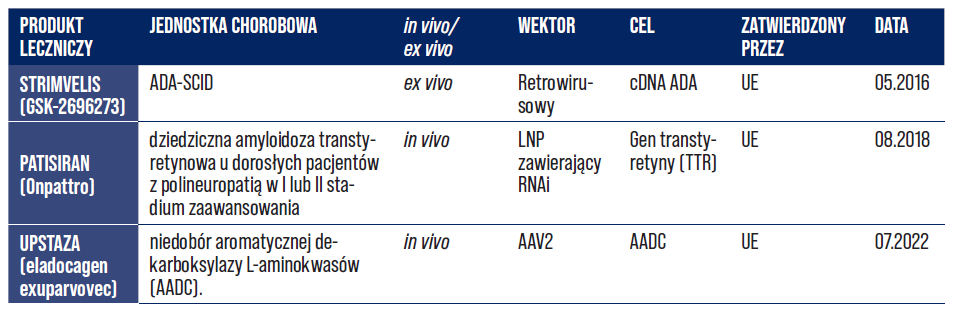
Tabela 5. Zestawienie produktów leczniczych terapii genowej zatwierdzonych wyłącznie w USA [33].
Wirus związany z adenowirusem
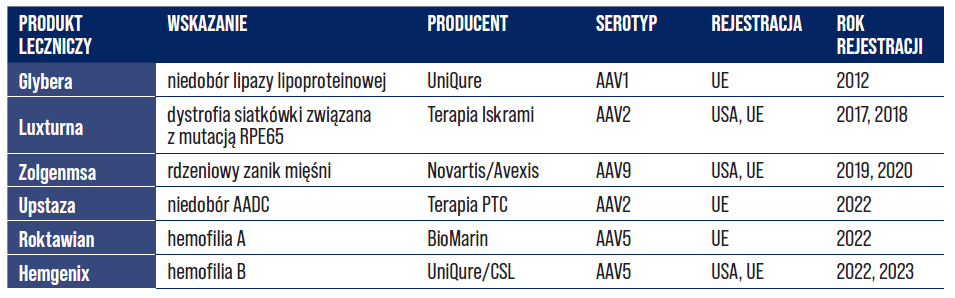
Wirus związany z adenowirusem (ang. adeno-associated virus; AAV) to mały, niepatogenny i niezdolny do replikacji wirus, który zakaża niektóre gatunki ssaków naczelnych, w tym także ludzi. AAV jest zbudowany z kapsydu białkowego oraz liniowego jednoniciowego DNA, który koduje geny niezbędne dla jego cyklu życiowego. Rekombinowany AAV, który jest wykorzystywany jako wektor do dostarczania transgenów terapeutycznych, rzadko integruje się z genomem komórki gospodarza, funkcjonuje głównie jako episom. [21-24]. Obecnie AAV jest głównym sposobem dostarczania terapii genowej in vivo. Pierwszą zatwierdzoną terapią genową z wykorzystaniem AVV był lek Glybera, stosowany w terapii niedoboru lipazy lipoproteinowej (ang. lipoprotein lipase deficiency, LPLD). Został on jednak wycofany z rynku w 2017 r. Od tego czasu w Stanach Zjednoczonych i Unii Europejskiej (UE) zatwierdzono łącznie aż pięć kolejnych terapii genowych opartych na AAV, z czego trzy w samym 2022 r. (Tabela 6).
Tabela 6. Produkty lecznicze terapii genowej zatwierdzone w UE oraz USA.
Ze względu na stosunkowo niską immunogenność, toksyczność, a także trwałą skuteczność oraz szeroki tropizm AAV stanowi bardzo obiecujący środek w terapii wielu chorób, w tym chorób ośrodkowego układu nerwowego (OUN), oczu, mięśni czy wątroby [25]. Jednakże ostatnio odnotowano przypadki ciężkiej toksyczności, a nawet śmierci pacjentów [24, 26], które wzbudziły obawy dotyczące bezpieczeństwa terapii genowej z wykorzystaniem AAV. Opracowano wiele metod ograniczania i minimalizowania wpływu odpowiedzi immunologicznych przeciwko kapsydom AAV lub produktom transgenicznym. Przedmiotowe metody obejmują m.in. zastosowanie środków immunosupresyjnych, pułapek kapsydowych, plazmaferezy, proteazy IgG czy indukcję regulatorowych limfocytów T [24, 27]. Niemniej jednak odpowiedź immunologiczna wciąż stanowi główne wyzwanie w terapii genowej AAV, ograniczając kwalifikację pacjentów, zwiększając ryzyko niebezpieczeństwa oraz zmniejszając skuteczność zastosowanej terapii. Ponadto w przypadku AAV niska skuteczność dostarczania, zwłaszcza do OUN, ze względu na barierę krew-mózg (ang. blood–brain barier, BBB), pozostaje poważnym wyzwaniem dla klinicznego zastosowania tej terapii genowej. Zatem istnieje pilna potrzeba wykorzystania inżynierii AAV do odkrycia kapsydów nowej generacji o ulepszonych właściwościach, np. zwiększonej penetracji BBB czy niższej immunogenności [24]. U pacjentów, którzy przyjęli lek oparty na wirusie z klasy AAV, wytwarza się trwała odporność. W związku z powyższym leki te podaje się tylko raz. Niestety część osób ma naturalnie nabytą odporność na ten typ wirusa – zostało to odnotowane u kilku procent niemowląt oraz u aż 40–50% osób dorosłych. U takich osób nie można zastosować terapii opartej na wirusie AAV [28].
Obecnie można wyróżnić następujące terapie genowe oparte na wirusie AAV:
- Produkt leczniczy Luxturna, który zawiera w swoim składzie woretygen neparwowek, zmodyfikowany wirus, który posiada prawidłową kopię genu RPE65. Wskazania do stosowania niniejszej terapii genowej obejmują leczenie pacjentów dorosłych oraz dzieci, którzy stracili wzrok z powodu dziedzicznej dystrofii siatkówki wywołanej przez mutacje genu RPE65. Białko nabłonka barwnikowego siatkówki o masie 65 kilodaltonów (RPE65) przekształca all-trans-retinol w 11-cis-retinol. Natomiast 11-cis-retinol tworzy chromofor podczas cyklu widzenia. Powyższe etapy stanowią punkt krytyczny dla biologicznej konwersji fotonu światła w sygnał elektryczny w obrębie siatkówki. Na skutek mutacji genu RPE65 następuje zmniejszenie aktywności lub zahamowanie aktywności izomerazy RPE65 all-trans-retinylowej, co w następstwie prowadzi do utraty wzroku, a ostatecznie doprowadza do ślepoty. Na skutek wstrzyknięcia woretygenu neparwowek do przestrzeni podsiatkówkowej następuje transdukcja cDNA kodującego prawidłowe ludzkie białko RPE65 do komórek nabłonka barwnikowego zlokalizowanych w siatkówce, co daje możliwość przywrócenia cyklu widzenia. Przeciwwskazania do zastosowania woretygenu neparwowek obejmują nadwrażliwość na substancję czynną lub substancję pomocniczą, zakażenie wewnątrz gałki ocznej lub zakażenie okołogałkowe, a także czynne zapalenie wewnątrzgałkowe [29-31].
- Onasemnogen abeparwowek stanowi produkt leczniczy terapii genowej o nazwie Zolgensma. Onasemnogen abeparwowek to niereplikujący rekombinowany wektor, który jest oparty na wirusie związanym z adenowirusami serotypu 9 (ang. adeno-associated virus serotype 9, AAV9-based vector). Zawiera on cDNA ludzkiego genu odpowiedzialnego za przeżycie neuronów ruchowych (ang. survival motor neuron, SMN), który jest kontrolowany hybrydowym promotorem wzmacniacza cytomegalowirusa / promotora beta-aktyny kurczaka, co zapewnia ciągłą i stale utrzymującą się ekspresję białka SMN. Mając na uwadze, że onasemnogen abeparwowek stanowi alternatywne źródło do ekspresji białka SMN w neuronach ruchowych, oczekuje się, że po podaniu będzie on promował przetrwanie oraz funkcjonowanie transdukowanych neuronów ruchowych. W przeprowadzonych dotychczas badaniach wykazano możliwość przedostawania się kapsydu AAV9 przez barierę krew-mózg oraz transdukowania neuronów ruchowych. Metoda rekombinacji DNA jest wykorzystywana do wytwarzania onasemnogenu abeparwowek w komórkach embrionalnych nerki [32]. Preparat Zolgensma podawany jest jednorazowo we wlewie dożylnym, który trwa ok. 1 godziny. Dawka jest przeliczana odpowiednio do wagi pacjenta. Największa możliwa do przyjęcia dawka może zostać zastosowana u pacjenta o masie ciała 21 kg [28, 32]. Produkt Zolgensma jest obecnie stosowany w leczeniu dzieci z rdzeniowym zanikiem mięśni (ang. spinal muscular atrophy, SMA) 5q z bialleliczną mutacją genu SMN1 oraz klinicznym rozpoznaniem SMA typu 1, a także w terapii pacjentów z rdzeniowym zanikiem mięśni 5q z bialleliczną mutacją genu SMN1 i z nie więcej niż 3 kopiami genu SMN2. Preparat ten stosuje się w terapii niemowląt oraz małych dzieci, u których objawy SMA jeszcze nie występują bądź też pojawiły się stosunkowo niedawno – utrata neuronów postępuje z dnia na dzień, a zapotrzebowanie na białko SMN jest najwyższe. Jedynym wyszczególnionym przez producenta przeciwwskazaniem do zastosowania onasemnogenu abeparwowek jest nadwrażliwość na substancję czynną lub na którąkolwiek substancję pomocniczą. Warto również zwrócić uwagę, iż przed rozpoczęciem terapii lekiem Zolgensma należy wykonać szczegółowe badanie krwi w celu dokonania oceny czynności wątroby. Należy także podkreślić, iż wyższe dawki produktu leczniczego są proporcjonalnie bardziej szkodliwe. Po zakończeniu terapii zalecane jest dalsze monitorowanie enzymów wątrobowych przez okres co najmniej 3 miesięcy [32, 33].
- Substancją aktywną produktu leczniczego terapii genowej o nazwie Upstaza jest eladokagen eksuparwowek. Eladokagen eksuparwowek to niereplikujący się, rekombinowany wektor wirusowy, który jest związany z adenowirusami serotypu 2
(ang. adeno-associated virus serotype 2; AAV2 based vector). Ten rekombinowany wektor wirusowy zawiera cDNA ludzkiego genu, który koduje enzym DOPA-dekarboksylazy (DDC) pod kontrolą promotora genów natychmiastowych wczesnych cytomegalowirusa. Przedmiotowy produkt leczniczy terapii genowej aktywuje ekspresję ludzkiego enzymu – dekarboksylazy L-aminokwasów aromatycznych (hAADC), a w następstwie dochodzi do wytwarzania dopaminy, a tym samym rozwoju funkcji motorycznych u pacjentów z niedoborem AADC. Niedobór AADC stanowi wrodzony błąd w biosyntezie neuroprzekaźnika z dziedziczeniem autosomalnym recesywnym w genie DDC, który koduje enzym AADC. Wspomniany enzym posiada zdolność do przekształcania L-3,4-dihydroksyfenyloalaniny (L-DOPA) w dopaminę. Mutacja genu DDC prowadzi do zmniejszenia lub zahamowania aktywności enzymu AADC, a zatem do zmniejszenia stężenia dopaminy, a to w następstwie skutkuje brakiem osiągnięcia etapów rozwoju przez większość pacjentów z niedoborem AADC. W komórkach embrionalnych nerki wykorzystywana jest metoda rekombinacji DNA do wytwarzania eladokagenu eksuparwowek. Produkt Upstaza został zarejestrowany na terenie UE oraz Wielkiej Brytanii w leczeniu pacjentów w wieku 18 miesięcy oraz starszych z klinicznie, molekularnie i genetycznie potwierdzonym rozpoznaniem niedoboru AADC z ciężkim fenotypem. Przeciwwskazania do zastosowania eladokagenu eksuparwoweku obejmują nadwrażliwość na substancję czynną lub substancję pomocniczą [33, 34]. - Kolejny produkt leczniczy terapii genowej posiada nazwę handlową Roctavian. Jego substancją czynną jest waloktokogen roksaparwoweku, który jest wskazany do stosowania w leczeniu ciężkiej postaci hemofilii typu A (wrodzony niedobór czynnika VIII) u osób dorosłych, u których nie występowały wcześniej inhibitory czynnika VIII, a także u których nie wykryto przeciwciał przeciwko serotypowi 5 adenowirusa (ang. adeno-associated virus serotype 5; AAV5 based vector). Waloktokogen roksaparwoweku jest niereplikującym się, rekombinowanym wektorem opartym na serotypie wirusa towarzyszącego adenowirusom AAV. Wektor ten zawiera cDNA postaci SQ pozbawiony domeny B genu ludzkiego czynnika krzepnięcia VIII pod kontrolą promotora specyficznego dla wątroby. Na skutek podania powoduje ekspresję postaci SQ pozbawionego domeny B ludzkiego czynnika krzepnięcia VIII (hFVIII-SQ). Waloktokogen roksaparwoweku jest wytwarzany z wykorzystaniem techniki rekombinowanego DNA z zastosowaniem systemu ekspresji bakulowirusa, który jest izolowany z komórek Spedoptera frugiperda (linia komórek Sf9). Przeciwwskazania do zastosowania waloktokogenu roksaparwoweku obejmują nadwrażliwość na substancję czynną lub substancje pomocnicze oraz aktywne zakażenia, zarówno te ostre, jak również przewlekłe niekontrolowane. Przeciwwskazaniem do zastosowania terapii genowej jest również istotne włóknienie oraz marskość wątroby [33, 35].
- Hemgenix (etranakogen dezaparwoweku) to produkt leczniczy terapii genowej. wykazujący ekspresję IX ludzkiego czynnika krzepnięcia. Etranakogen dezaparwoweku to niereplikujący, rekombinowany wektor oparty na wirusie związanym z adenowirusami serotypu 5, który zawiera DNA o zoptymalizowanych kodonach genu ludzkiego czynnika krzepnięcia IX wariantu R338L (FIX-Padwa) znajdujący się pod kontrolą promotora specyficznego dla wątroby (LP1). Wytwarzanie etranakogenu dezaparwoweku następuje z wykorzystaniem metody rekombinacji w komórkach owadów. Wskazania do włączenia leczenia preparatem Hemgenix obejmują ciężką i umiarkowanie ciężką postać hemofilii B (wrodzony niedobór czynnika IX) u dorosłych pacjentów, u których nie występowały wcześniej inhibitory czynnika IX. Na skutek jednorazowej infuzji dożylnej przedmiotowego produktu leczniczego rekombinowany wektor preferencyjnie dociera do komórek wątroby, gdzie jego DNA znajduje się wyłącznie w postaci episomalnej. Na skutek transdukcji etranakogen dezaparwoweku wywołuje długoterminową, specyficzną dla wątroby ekspresję białka czynnika IX-Padwa. W związku z powyższym etranakogen dezaparwoweku może częściowo lub całkowicie łagodzić niedobór aktywności prokoagulacyjnej krążącego czynnika IX u pacjentów ze zdiagnozowaną hemofilią B. Przeciwwskazania do zastosowania etranakogenu dezaparwoweku obejmują nadwrażliwość na substancję czynną lub substancje pomocnicze oraz aktywne zakażenia, zarówno te ostre, jak również przewlekłe niekontrolowane. Przeciwwskazaniem do zastosowania terapii genowej jest również istotne włóknienie oraz marskość wątroby [33, 36].
Terapie CAR-T
W ostatniej dekadzie terapia komórkami CAR-T stanowi innowacyjne, obiecujące podejście w leczeniu nowotworów hematologicznych [37]. Do tej pory przeprowadzono aż 105 badań klinicznych z wykorzystaniem komórek CAR-T (16 z nich jest obecnie w fazie III badań klinicznych). W ciągu ostatnich 5 lat EMA zatwierdziła pierwsze 6 produktów CAR-T ukierunkowanych na antygen CD19 lub B (BCMA) [38]. Kolejne wskazania czynią z CAR-T alternatywę dla dotychczasowych terapii – obecnie są prowadzone badania w kierunku zastosowania tej metody w nowotworach hematologicznych oraz guzach litych. Dalsze potencjalne cele terapeutyczne wychodzą poza onkologię, gdyż dotyczą SMA, HIV czy Covid-19.
W pierwszej fazie mechanizm terapii z wykorzystaniem komórek CAR-T obejmuje izolowanie limfocytów T z krwi obwodowej bądź też drogą leukaferezy. W kolejnym etapie oddziela się limfocyty Th17, a ich ilość ulega zwiększeniu poprzez wymuszone namnażanie limfocytów T w warunkach ex vivo. Dalej następuje ich transfekcja, a zatem wprowadzenie informacji genetycznej dotyczącej wyglądu przyszłej chimery przeciwciała z receptorem oraz rodzaju antygenu za pomocą wektora wirusowego (lentiwirusa lub retrowirusa) do wspomnianych komórek, co umożliwi im rozpoznawanie komórek nowotworowych. Zatem w wyniku zmian genetycznych zachodzących z wykorzystaniem wirusa wektorowego pozbawionego wirulencji w limfocytach T powstaje receptor komórki T (ang. T cell receptor, TCR), którego domena zewnątrzkomórkowa jest zastąpiona fragmentem przeciwciała, które jest swoiście skierowane przeciwko antygenowi nowotworowemu (scFv). Przeprowadzenie powyższych modyfikacji prowadzi do uzyskania limfocytów T z chimerycznym receptorem antygenowym (CAR), tzw. CAR-T. Ich nazwa obejmuje pojęcie „chimeryczny”, gdyż łączy w sobie dwa rodzaje odpowiedzi swoistej układu immunologicznego – cytotoksycznej (limfocyty T) oraz humoralnej (przeciwciała). W ostatnim etapie komórki efektorowe są namnażane, a następnie z powrotem są podawane pacjentowi in venam [39, 40]. W wyniku powyższego następuje aktywacja zstępującej kaskady sygnałowej, co w następstwie prowadzi do aktywacji i proliferacji limfocytów T, a także do nabycia przez nie funkcji efektorowych i wydzielania zapalnych cytokin i chemokin. Sekwencja powyższych zdarzeń prowadzi do wywołania apoptozy i nekrozy docelowych komórek prezentujących antygen CD19 lub B (BCMA) [41].
Obecnie można wyróżnić następujące terapie CAR-T:
- Preparat o nazwie handlowej Abecma (idekabtagen wikleucel). Wskazanie do stosowania obejmuje dorosłych pacjentów z nawrotowym oraz opornym na leczenie szpiczakiem plazmocytowym, którzy otrzymali już co najmniej trzy linie terapii (środek immunomodulujący, inhibitor proteasomu, przeciwciało anty-CD38) i w wyniku których wykazali progresję choroby podczas zastosowanej ostatniej linii leczenia.
Ta zmodyfikowana genetycznie autologiczna immunoterapia jest złożona z ludzkich limfocytów T transdukowanych wektorem lentiwirusowym (ang. lentiviral vector, LVV) kodującym chimeryczny receptor antygenowy (ang. chimeric antigen receptor, CAR) rozpoznający antygen dojrzewania limfocytów B (ang. B-cell maturation antygen, BCMA). Wspomniany antygen ulega ekspresji na powierzchni zarówno zdrowych, jak i złośliwych komórek plazmatycznych. Chimeryczny receptor antygenowy posiada aż cztery domeny: domenę przeciwko antygenowi dojrzewania limfocytów B ukierunkowaną na scFv w celu nadania swoistości wobec antygenu, domenę transbłonową, domenę CD3-zeta (CD3ζ) aktywującą limfocyty T oraz domenę ko-stymulującą 4-1BB. W wyniku podania idekabtagenu wikleucelu następuje aktywacja limfocytów ukierunkowanych na określone antygeny, co przyczynia się do namnażania się limfocytów T z ekspresją receptora CAR, a także wydzielania cytokin. W następstwie aktywność cytolityczna niszczy komórki z ekspresją BCMA. Idecabtagene vicleucel to pierwsza zatwierdzona terapia z wykorzystaniem komórek CAR-T ukierunkowana na rozpoznawanie i wiązanie BCMA – białka, które ulega ekspresji na większości komórek nowotworowych szpiczaka plazmocytowego. Pacjenci z współistniejącym czynnym zaburzeniem ośrodkowego układu nerwowego (OUN), nieprawidłową czynnością wątroby, płuc nerek, czy serca są bardziej narażeni na występowanie działań niepożądanych i wymagają oni szczególnej uwagi [33, 42]. - Preparat Carvykti (ciltakabtagen autoleucel) to kolejna ukierunkowana na BCMA, genetycznie zmodyfikowana autologiczna immunoterapia limfocytami T transdukowanymi ex vivo za pomocą niekompetentnego replikacyjnie wektora lentiwirusowego, kodującego CAR. BCMA ulega ekspresji głównie na powierzchni komórek linii B szpiczaka mnogiego, jak również komórek B i komórek plazmatycznych będących w późnym stadium rozwoju. Białko CAR zostało tak zaprojektowane, by zapewnić wysoką reaktywność przeciwko BCMA. Po związaniu się z komórkami wykazującymi ekspresję BCMA CAR promuje aktywację limfocytów T, ekspansję i eliminację komórek docelowych. Ciltakabtagen autoleucel jest wskazany do leczenia dorosłych pacjentów, u których zdiagnozowano nawrotowego i opornego na leczenie szpiczaka mnogiego oraz stosowano już minimum trzy metody terapii (lek immunomodulujący, inhibitor proteasomu i przeciwciało anty-CD38) i stwierdzono progresję choroby w trakcie przeprowadzenia ostatniej metody leczenia.
- Kolejnym preparatem jest produkt leczniczy Breyanzi (lisocabtagene maraleucel). Chłoniaki agresywne z dużych komórek B stanowią rozbudowaną grupę nowotworów, które wywodzą się z dojrzałych, obecnych we krwi obwodowej limfocytów B. Najczęściej diagnozowany jest chłoniak rozlany z dużych komórek B (ang. diffuse large B-cell lymphoma, DLBCL) [39]. Przedmiotowy produkt został zarejestrowany przez FDA i EMA w 3 wskazaniach obejmujących różne podtypy LBCL, tj. nawrotowy lub oporny na leczenie chłoniak rozlany z dużych komórek B (DLBCL), pierwotny chłoniak śródpiersia z dużych komórek B (ang. primary mediastinal B-cell lymphoma, PMBCL) oraz chłoniak grudkowy stopnia 3B (ang. follicular lymphoma grade 3B, FL3B). Ta immunoterapia jest włączana do leczenia dorosłych pacjentów z powyższymi jednostkami chorobowymi po zastosowaniu dwóch lub więcej linii leczenia systemowego. Lisocabtagene maraleucel nie jest wskazany w leczeniu pacjentów z pierwotnym chłoniakiem ośrodkowego układu nerwowego. Immunoterapia ta jest ukierunkowana na antygen CD19, wykorzystując zmodyfikowane genetycznie autologiczne komórki. Produkt Breyanzi zawiera oczyszczone limfocyty T CD8+ i CD4+, które są podawane o ustalonym składzie w celu zmniejszenia zmienności dawki tych limfocytów. Chimeryczny receptor antygenowy CAR zawiera jednołańcuchowy fragment zmienny (ang. single-chain variable fragment, scFv), który jest pozyskany z mysiego przeciwciała monoklonalnego FMC63, a także region zawiasowy IgG4, domenę przezbłonową CD28, domenę kostymulacyjną 4-1BB (CD137) i domenę aktywacyjną zeta CD3. Sygnalizacja z wykorzystaniem domeny zeta CD3 stanowi punkt krytyczny dla inicjacji aktywacji limfocytów T, a także dla aktywności przeciwnowotworowej. Natomiast za zwiększone rozprzestrzenianie i utrzymanie immunoterapii odpowiedzialna jest domena 4-1BB (CD137). Na skutek związania się receptora CAR z antygenem CD19, który ulega ekspresji na powierzchni zmienionych nowotworowo i prawidłowych limfocytów B, następuje indukcja aktywacji i proliferacji limfocytów T z ekspresją receptora CAR oraz rozpoczyna się uwalnianie cytokin prozapalnych. W następstwie zastosowania immunoterapii dochodzi do cytotoksycznego uśmiercania komórek docelowych [33, 43].
- Następną autologiczną immunokomórkową terapią przeciwnowotworową jest produkt leczniczy Kymriah. Zawiera on w swoim składzie tisagenlecleucel – limfocyty T zmodyfikowane genetycznie w warunkach ex vivo przy użyciu wektora lentiwirusowego kodującego chimeryczny receptor antygenowy anty-CD19 w celu wykrycia i wyeliminowania komórek z ekspresją CD19. Tisagenlecleucel jest wskazany w terapii dzieci, młodzieży oraz młodych dorosłych przed ukończeniem 25 r.ż. z ostrą białaczką limfoblastyczną (ang. acute lymphoblastic leukaemia, ALL) z komórek B, oporną na leczenie, znajdującą się w fazie nawrotu po transplantacji bądź w fazie co najmniej drugiego nawrotu. Dodatkowo wskazania do stosowania wspomnianego produktu leczniczego obejmują także dorosłych z nawracającym lub opornym na leczenie DLBCL, po przebyciu co najmniej dwóch linii leczenia systemowego [33, 44].
- Produkt leczniczy Yescarta zawiera aksykabtagen cyloleucel, a zatem genetycznie zmodyfikowane autologiczne limfocyty T transdukowane ex vivo przy zastosowaniu wektora retrowirusowego prezentującego CAR anty-CD19 zawierający ScFv anty-CD19 powiązany zarówno z domeną kostymulującą CD-28, jak również z domeną sygnalizacyjną CD3ζ. Na skutek wdrożenia przedmiotowej immunoterapii u pacjentów następuje związanie nie tylko z komórkami nowotworowymi prezentującymi CD19,
ale również ze zdrowymi limfocytami B genetycznie zmodyfikowanych autologicznych limfocytów T. W wyniku powyższego następuje aktywacja zstępującej kaskady sygnałowej, co prowadzi do aktywacji i proliferacji limfocytów T, a także do nabycia przez nie funkcji efektorowych i wydzielania zapalnych cytokin i chemokin. Ta sekwencja zdarzeń prowadzi do apoptozy i nekrozy docelowych komórek prezentujących CD19. Produkt leczniczy Yescarta jest wskazany w terapii dorosłych pacjentów ze zdiagnozowanym DLBCL. Produkt ten jest również wykorzystywany w leczeniu pacjentów z progresją chłoniaka o wysokim stopniu złośliwości z komórek B (ang. high-grade B-cell lymphoma, HGBL) w ciągu 12 miesięcy od zakończenia prowadzenia chemioimmunoterapii pierwszego rzutu bądź w przypadku oporności na chemioterapię. Wskazania do stosowania aksykabtagen cyloleucel obejmują także terapię nawrotowego lub opornego na leczenie DLBCL oraz PMBCL u dorosłych pacjentów, którzy zostali poddani minimum dwóm liniom leczenia systemowego [33, 44, 45]. - Breksukabtagen autoleucel to kolejne genetycznie zmodyfikowane autologiczne limfocyty T transdukowane ex vivo przy użyciu wektora retrowirusowego prezentującego CAR anty-CD19 zawierający mysi scFv anty-C19 bezpośrednio powiązany z domeną kostymulującą CD28, jak i domeną sygnalizacyjną CD3-zeta. Są one dostępne w postaci produktu leczniczego o nazwie Tecartus, który jest stosowany w immunoterapii skierowanej przeciwko CD19. Breksukabtagen autoleucel stosuje się w leczeniu dorosłych pacjentów z nawrotowym lub opornym na leczenie chłoniakiem z komórek płaszcza (ang. mantle cell lymphoma, MCL), którzy otrzymali co najmniej dwie linie leczenia systemowego. Niniejszy produkt leczniczy jest również wskazany w leczeniu nawrotowej lub opornej na leczenie ALL, która wywodzi się z prekursorów limfocytów B u dorosłych pacjentów powyżej 26 r.ż. [33, 46].
Przeciwwskazania do zastosowania terapii CAR-T
W przypadku przeciwskazań do zastosowania chemioterapii limfodeplecyjnej przeciwskazane jest również zastosowanie terapii CAR-T u pacjenta. Przeciwskazania do zastosowania chemioterapii limfodeplecyjnej obejmują, m.in. ostre zakażenia, aplazję szpiku lub zahamowanie czynności szpiku przed rozpoczęciem leczenia, zakażenie dróg moczowych, ostre zmiany w obrębie nabłonka przejściowego w wyniku chemioterapii cytotoksycznej i radioterapii, obturację odpływu moczu, zaburzenia czynności nerek, kiedy klirens kreatyniny wynosi <30 ml/min, niewyrównaną niedokrwistość hemolityczna oraz karmienie piersią. Ponadto przeciwwskazania do stosowania immunoterapii z wykorzystaniem limfocytów T z ekspresją receptora CAR obejmują aktywne lub utajone zakażenie HBV, aktywne zakażenie HCV lub HIV, ostrą lub przewlekłą intensywną chorobę przeszczep-przeciw-gospodarzowi (ang. graft-versus-host disease, GVHD) oraz ciążę. W przypadku odnotowania któregokolwiek z powyższych przeciwwskazań infuzję limfocytów T z ekspresją receptora CAR należy opóźnić, aby uniknąć potencjalnie ciężkiej aktywacji immunologicznej oraz związanych z nią powikłań [38].
Terapia genowa z wykorzystaniem komórek macierzystych
Podstawą niniejszej terapii genowej są komórki macierzyste (komórki CD34+) pochodzące z własnego szpiku kostnego lub krwi pacjenta, które zostały zmodyfikowane genetycznie w celu zamieszczenia kopii genu, w wyniku czego mogą się one dzielić, co prowadzi do wytworzenia innych rodzajów komórek krwi [47].
- Pierwszym przykładem niniejszej terapii genowo-komórkowej jest produkt leczniczy o nazwie Libmeldy (atidarsagen autotemcel), który zawiera zmodyfikowane genetycznie w warunkach ex vivo autologiczne komórki krwiotwórcze i progenitorowe (HSPC) CD34+. Te autologiczne HSPC CD34+ pobierane są z krwi obwodowej pacjenta, a następnie są transdukowane wektorem lentiwirusowym (ARSA LVV), co powoduje insercję jednej lub większej liczby kopii ludzkiego komplementarnego kwasu deoksyrybonukleinowego (cDNA) kodującego ARSA do genomu komórki. Dzięki przeprowadzonej modyfikacji genetycznej komórki stają się zdolne do ekspresji funkcjonalnego enzymu ARSA. Atidarsagen autotemcel jest stosowany w leczeniu leukodystrofii metachromatycznej (ang. metachromatic leukodystrophy, MLD), która odznacza się dwuallelowymi mutacjami genu arylosulfatazy A prowadzącymi do zmniejszenia aktywności enzymatycznej ARSA. Produkt leczniczy terapii genowo-komórkowej stosuje się zarówno w terapii dzieci z późnymi niemowlęcymi, jak również wczesnymi młodzieńczymi postaciami choroby, bez odnotowanych objawów klinicznych. Libmeldy stosuje się również u dzieci z wczesną młodzieńczą postacią choroby, u których występują wczesne objawy kliniczne, jednakże przed pojawieniem się zaburzeń funkcji poznawczych oraz zaburzeń samodzielnego poruszania się. Przeciwwskazaniem do zastosowania terapii genowo-komórkowej z wykorzystaniem produktu leczniczego Libmeldy jest nadwrażliwość na substancję czynną lub na substancję pomocniczą oraz wcześniejsze leczenie terapią genową z krwiotwórczymi komórkami macierzystymi. Produktu leczniczego Libmeldy nie można zastosować u pacjentów, u których odnotowano przeciwwskazania do stosowania produktów leczniczych do mobilizacji i mieloablacji [33, 48].
- Kolejnym produktem leczniczym terapii genowo-komórkowej jest Strimvelis, który jest wskazany w terapii pacjentów z ciężkim, złożonym niedoborem odporności spowodowanym niedoborem deaminazy adenozynowej (ADA-SCID), dla których nie jest dostępny odpowiedni dawca komórek macierzystych dopasowany pod względem antygenu ludzkich leukocytów (HLA). Jest to autologiczna frakcja komórek wzbogacona o CD34. Komórki CD34+ są transdukowane wektorem retrowirusowym posiadającym ludzką sekwencję cDNA deaminazy adenozynowej (ADA) z ludzkich komórek macierzystych układu krwiotwórczego/progenitorowych (CD34+). Po przeprowadzeniu infuzji, komórki CD34+ ulegają wszczepieniu do szpiku kostnego, gdzie w następstwie wywołują repopulację układu krwiotwórczego komórkami wytwarzającymi enzym ADA w stężeniach aktywnych farmakologicznie. Po przeprowadzeniu infuzji działanie produktu powinno utrzymywać się przez całe życie. Przeciwwskazaniem do zastosowania Strimvelis jest nadwrażliwość na substancję czynną lub na substancje pomocnicze. Ponadto przeciwwskazania do zastosowania przedmiotowej terapii genowo-komórkowej obejmują białaczkę, mielodysplazję (zarówno obecnie występująca bądź w przeszłości), przyjmowanie terapii genowo-komórkowej w przeszłości oraz dodatni wynik badania w kierunku zakażenia ludzkim wirusem upośledzenia odporności (HIV), jak również każdym innym czynnikiem wymienionym w dyrektywie UE w sprawie komórek i tkanek [33, 49].
Terapia genowa oparta na technologii CRISPR-Cas9
W 2023 r. niezwykle ważnym wydarzeniem w rozwoju światowej medycyny było zatwierdzenie przez FDA pierwszej terapii opartej na technologii CRISPR-Cas9 umożliwiającej precyzyjną edycję DNA w obszarze zmutowanych genów [50]. Od lutego 2024 r. został on również zarejestrowany w UE. Produkt leczniczy terapii genowej nosi nazwę handlową Casgevy i jest on stosowany w terapii pacjentów cierpiących na anemię sierpowatokrwinkową (ang. sickle cell disease, SCD). Produkt leczniczy Casgevy to terapia komórkowa zawierająca autologiczne krwiotwórcze komórki macierzyste i komórki HSPC ex vivo CD34+ zmodyfikowane genetycznie przy użyciu technologii CRISPR/Cas9. Swoisty RNA naprowadzający pozwala na uzyskanie za pomocą technologii CRISPR/Cas9 pęknięcia dwuniciowego DNA w miejscu wiązania czynnika transkrypcyjnego GATA1 w swoistym dla komórek linii erytroidalnej regionie sekwencji wzmacniającej genu BCL11A. Na skutek powyższych modyfikacji następuje nieodwracalne zakłócenie wiązania czynnika transkrypcyjnego GATA1 i tym samym spadek ekspresji genu BCL11A, który przyczynia się do nasilenia ekspresji γ-globiny oraz powstawania hemoglobiny płodowej (HbF) w komórkach linii erytroidalnej. Jest to bezpośrednia odpowiedź na przyczynę, która leży u podstawy rozwoju dwóch jednostek chorobowych – brak globiny w β-talasemii zależnej od przetoczeń (TDT) oraz nieprawidłowa globina w SCD. Zastosowanie przedmiotowej terapii genowej w β-talasemii może doprowadzić do ograniczenia nieefektywnej erytropoezy i hemolizy, a także do wzrostu stężenia hemoglobiny całkowitej. Natomiast u pacjentów z ciężką postacią SCD na skutek ekspresji HbF dochodzi do obniżenia wewnątrzkomórkowego stężenia HbS, co może zapobiegać powstawaniu sierpowatych krwinek czerwonych. Przeciwwskazaniem do zastosowania przedmiotowej terapii jest nadwrażliwość na substancję czynną lub na substancje pomocnicze. Produktu leczniczego Casgevy nie można zastosować u pacjentów, u których odnotowano przeciwwskazania do stosowania produktów leczniczych do mobilizacji i mieloablacji [33, 51].
PODSUMOWANIE
Miniony rok 2023 został uznany za przełomowy w dziedzinie zarówno terapii genowych, jak i komórkowych ze względu na liczbę dopuszczonych do obrotu nowych terapii genowych,
jak również z powodu zatwierdzenia pierwszej terapii genowej, która jest oparta na przełomowej technologii CRISPR/Cas9. Terapia genowa stanowi wszechstronne narzędzie, które może przyspieszyć rozwój medycyny spersonalizowanej, rewolucjonizując skuteczność terapii, a także łagodząc skutki uboczne stosowanych leków. Największy rozwój medycyny spersonalizowanej można zaobserwować w obszarze leczenia nowotworów i chorób dziedzicznych, gdzie strategie i produkty terapii genowej odgrywają kluczową rolę. Jednakże potencjał terapii genowej wykracza daleko poza powyższe domeny i obejmuje także różnorodne patologie, takie jak dolegliwości neurodegeneracyjne, zaburzenia immunologiczne czy procesy zapalne. W związku z powyższym na nadchodzące lata zostały rozbudzone wysokie oczekiwania w zakresie poszukiwania nowych terapii genowych. Niewątpliwie ciągłe badania i innowacje w tej dziedzinie mogą doprowadzić do rozwoju skuteczniejszych metod leczenia wielu jednostek chorobowych, co tym samym może zmienić życie wielu pacjentów na całym świecie.
dr n. farm. Agnieszka Zajda
Uniwersytet Medyczny w Łodzi
Piśmiennictwo
1. Nawrocki S., Mackiewicz A. Terapia genowa nowotworów wyzwaniem XXI wieku. Współczesna Onkologia. 2000, 4, 190–194.
2. Arjmand B., Larijani B., Sheikh Hosseini M., Payab M., Gilany K., Goodarzi P., Parhizkar Roudsari P., Amanollahi Baharvand M., Hoseini Mohammadi N. The Horizon of Gene Therapy in Modern Medicine: Advances and Challenges. Adv Exp Med Biol. 2020;1247:33-64.
3. Flotte T. Gene therapy: the first two decades and the current state-of-the-art. J Cell Physiol. 2007; 213; 301-305.
4. Culver K., Cornetta K., Morgan R., Morecki S., Aebersold P., Kasid A., Lotze M., Rosenberg S., Anderson W., Blaese R. Lymphocytes as cellular vehicles for gene therapy in mouse and man. Proc Natl Acad Sci USA. 1991, 88, 3155–3159.
5. Rosenberg S., Aebersold P., Cornetta K., Kasid A., Morgan R., Moen R., Karson E., Lotze M., Yang J., Topalian S. Gene transfer into humans—immunotherapy of patients with advanced melanoma, using tumor-infiltrating lymphocytes modified by retroviral gene transduction. N Engl J Med. 1990, 323, 570–578.
6 Blaese R., Culver K., Miller A., Carter C., Fleisher T., Clerici M., Shearer G., Chang L., Chiang Y., Tolstoshev P., Greenblatt J., Rosenberg S., Klein H,, Berger M,, Mullen C., Ramsey W., Muul L., Morgan R., Anderson W. T lymphocyte-directed gene therapy for ADA-SCID: Initial trial results after 4 years. Science, 1995, 270: 475–480.
7. Mateuszuk J. Perspektywy rozwoju terapii genowej w aspekcie zastosowania w chorobach uznawanych za nieuleczalne. Farmacja regionu północno-wschodniego. 2022, 4, 43-47.
8. Crystal R., McElvaney N., Rosenfeld M., Chu C., Mastrangeli A., Hay J., Brody S., Jaffe H., Eissa N., Danel C. Administration of an adenovirus containing the human CFTR cDNA to the respiratory tract of individuals with cystic fibrosis. Nat Genet. 1994, 8, 42–51.
9. Flotte T., Carter B., Conrad C., Guggino W., Reynolds T., Rosenstein B., Taylor G., Walden S., Wetzel R. A phase I study of an adeno-associated virus-CFTR gene vector in adult CF patients with mild lung disease. Hum Gene Ther. 1996, 7, 1145–1159.
10. Flotte T., Afione S., Conrad C., McGrath S., Solow R., Oka H., Zeitlin P., Guggino W., Carter B. Stable in vivo expression of the cystic fibrosis transmembrane conductance regulator with an adeno-associated virus vector. Proc Natl Acad Sci USA. 1993, 90, 10613–10617.
11. Kay M., Manno C., Ragni M., Larson P., Couto L., McClelland A., Glader B., Chew A., Tai S., Herzog R., Arruda V., Johnson F., Scallan C., Skarsgard E., Flake A., High K. Evidence for gene transfer and expression of factor IX in haemophilia B patients treated with an AAV vector. Nat Genet . 2000. 24, 257–261.
12. MacGregor R.. Clinical protocol. A phase 1 open-label clinical trial of the safety and tolerability of single escalating doses of autologous CD4 T cells transduced with VRX496 in HIV-positive subjects. Hum Gene Ther. 2001, 12, 2028–2029.
13. Glorioso J., Mata M., Fink D.. Gene therapy for chronic pain. Curr Opin Mol Ther. 2003, 5, 483–488.
14. Bank A., Dorazio R., Leboulch P. A phase I/II clinical trial of beta-globin gene therapy for beta-thalassemia. Ann NY Acad Sci. 2005, 1054, 308–316.
15. Levine B., Humeau L., Boyer J., MacGregor R., Rebello T., Lu X., Binder G., Slepushkin V., Lemiale F., Mascola J., Bushman F., Dropulic B., June C.. Gene transfer in humans using a conditionally replicating lentiviral vector. Proc Natl Acad Sci USA. 2006, 103, 17372–17377.
16. Mata M., Chattopadhyay M, Fink DJ. Gene therapy for the treatment of sensory neuropathy. Expert Opin Biol Ther. 2006, 6, 499–507.
17. Steffin D., Hsieh E., Rouce R. Gene therapy: Current applications and future possibilities. Adv Pediatr. 2019, 66, 37–54.
18. Kaufmann K., Buning H., Galy A., Schambach A., Grez M. Gene therapy on the move. EMBO Mol Med. 2013, 5, 1642–1661.
19. Thorne B., Takeya R., Vitelli F., Swanson X. Gene therapy. Adv Biochem Eng Biotechnol. 2018, 165, 351–399.
20. Gene Therapy Medicinal Products, https://www.pei.de/EN/medicinal-products/atmp/gene-therapy-medicinal-products/gene-therapy-node.html (stan z dnia 10.05.2024).
21. Vokinger K., Glaus C., Kesselheim A.. Approval and therapeutic value of gene therapies in the US and Europe. Gene Ther. 2023, 30, 756-760.
22. Samulski R., Muzyczka N. AAV-Mediated Gene Therapy for Research and Therapeutic Purposes. Annu Rev Virol. 2014, 1, 427-51.
23. Nakai H., Yant S., Storm T., Fuess S., Meuse L., Kay M. Extrachromosomal recombinant adeno-associated virus vector genomes are primarily responsible for stable liver transduction in vivo. J Virol. 2001, 75, 6969-76.
24. Ghauri M., Ou L. AAV Engineering for Improving Tropism to the Central Nervous System. Biology (Basel). 2023, 12, 186.
25. Mondal M., Guo J., He P., Zhou D. Recent advances of oncolytic virus in cancer therapy. Hum Vaccin Immunother. 2020,16, 2389-2402.
26. Shieh P., Bönnemann C., Müller-Felber W., Blaschek A., Dowling J., Kuntz N., Seferian A. Re: „Moving Forward After Two Deaths in a Gene Therapy Trial of Myotubular Myopathy” autorstwa Wilsona i Flotte. Szum. Gene The. 2020, 31, 787.
27 .Li X., Wei X., Lin J., Ou L. A versatile toolkit for overcoming AAV immunity. Front Immunol. 2022 Sep 2;13:991832.
28. Zolgensma® (terapia genowa), https://www.fsma.pl/leki/zolgensma/ (stan z dnia 15.05.2024).
29. Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Luxturna, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).
30. Beheshtizadeh N., Gharibshahian M., Pazhouhnia Z., Rostami M., Rajabi Zangi A., Maleki R., Kolahi Azar H., Zalouli V., Rajavand H., Farzin, A. Commercialization and regulation of regenerative medicine products: Promises, advances and challenges. Biomed. Pharmacother. 2022, 153, 113431.
31. Zhang W., Li L., Li D., Liu J., Li X., Li W., Xu X., Zhang M. Chandler L., Lin, H. The First Approved Gene Therapy Product for Cancer Ad-p53 (Gendicine): 12 Years in the Clinic. Hum. Gene Ther. 2018, 29, 160–179.
32. Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Zolgensma, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).
33. Shchaslyvyi, A., Antonenko, S., Tesliuk, M., Telegeev, G. Current State of Human Gene Therapy: Approved Products and Vectors. Pharmaceuticals 2023, 16, 1416.
34. Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Upstaza, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).
35.]Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Roctavian, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).
36. Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Hemgenix, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).
37. El-Kadiry, A., Rafei, M., Shammaa, R. Cell therapy: Types, regulation, and clinical benefits. Front. Med. 2021, 8, 756029.
38. European Medicines Agency, EU clinical trials register. 2020. https://www.clinicaltrialsregister.eu/ (stan z dnia 15.05.2024).
39. Wrona E., Potemski P. A novel immunotherapy — the history of CAR T-cell therapy. Oncol Clin Pract 2019; 15: 202–207.
40. Na czym polega terapia CAR-T w leczeniu nowotworów hematologicznych? https://www.onkonet.pl/dp_terapia_car-t.php (stan z dnia 15.05.2024).
41. Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Yescarta, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).
42. Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Carvykti, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).
43. Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Breyanzi, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).
44. Mahadeo K., Khazal S., Abdel-Azim H., Fitzgerald J., Taraseviciute A., Bollard C., Tewari P., Duncan C., Traube C., McCall D., Steiner M., Cheifetz I., Lehmann L., Mejia R., Slopis J., Bajwa R., Kebriaei P., Martin P., Moffet J., McArthur J., Petropoulos D., O'Hanlon Curry J., Featherston S., Foglesong J., Shoberu B., Gulbis A., Mireles M., Hafemeister L., Nguyen C., Kapoor N., Rezvani K., Neelapu S., Shpall E. Pediatric Acute Lung Injury and Sepsis Investigators (PALISI) Network. Management guidelines for paediatric patients receiving chimeric antigen receptor T cell therapy. Nat Rev Clin Oncol. 2019 Jan;16(1):45-63.
45. Shahryari A., Saghaeian Jazi M., Mohammadi S., Razavi Nikoo H., Nazari Z., Hosseini, E., Burtscher I., Mowla S., Lickert H. Development and Clinical Translation of Approved Gene Therapy Products for Genetic Disorders. Front. Genet. 2019, 10, 868.
46. Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Tecartus, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).
47. Libmeldy, https://www.ema.europa.eu/en/medicines/human/EPAR/libmeldy (stan z dnia 15.05.2024).
48. Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Libmeldy, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).
49. Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Strimvelis, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).
50. Co nowego w terapiach genowych? Podsumowanie 2023 roku. https://terapiegenowe.pl/artykuly/co-nowego-w-terapiach-genowych-podsumowanie-2023-roku/ (stan z dnia 15.05.2024).
51. Rejestr Produktów Leczniczych, Karta Charakterystyki Postaci Leku - Casgevy, https://rejestrymedyczne.ezdrowie.gov.pl/rpl/search/public (stan z dnia 12.05.2024).